 聚类分析中的机器学习与统计方法综述(一)
聚类分析中的机器学习与统计方法综述(一)

01
概况
单细胞转录组测序(scRNA-seq)技术能够对细胞群中的每一个细胞进行大规模的全转录组分析。它的核心分析是将单细胞聚类,以揭示细胞亚型,并根据细胞之间的关系推断细胞谱系。本文综述了在过去几年间发展起来的,用于单细胞转录组分析中聚类的机器学习和统计方法,重点介绍了如何将一些常见的聚类方法,如层次聚类、基于图的聚类、混合模型、k-means、集成学习、神经网络和基于密度的聚类等加以调整及应用,从而解决单细胞转录组数据分析中的独特挑战,例如低表达基因的缺失,转录本的不均匀覆盖,以及由技术偏差和不相关的混杂生物变异所带来的细胞标记的失真。我们评价了标准化、dropouts推测以及降维等预处理步骤如何提高聚类效果。此外,我们还将介绍一些能够对时间序列样本和多个细胞群进行聚类并且检测罕见细胞类型的新方法。最后,本文对部分开发用于单细胞转录组聚类分析的软件进行了实验和比较,以评估其性能和效率,为未来的数据分析提供一定的指导和方向。
02
介绍
细胞的转录组分析可以捕捉基因的表达活性,从而揭示细胞的身份和功能。在传统的bulk-RNA测序中,转录组是通过从生物样本中收集的大量细胞转录水平的平均值来测量的,这些平均后的表达值被用于基因共表达模块的识别和样本聚类。由于忽略了单个细胞的特性,这些传统的方法无法在单细胞分辨率上研究重要的生物学问题,如细胞在早期发育过程中的不同功能角色、复杂组织中的不同细胞类型和细胞谱系关系。目前,scRNA-seq技术已广泛用于量化单个细胞中的mRNA水平。在单细胞转录组的实验操作中,使用不同的捕获方法(如FACS,Fluidigm C1,microdroplet microfluidics)分离单细胞,然后对RNA进行逆转录并扩增测序。单细胞转录组的应用已经带来了重要的生物学见解和发现,例如,对癌症中肿瘤异质性的理解。
细胞聚类是单细胞转录组数据分析中识别细胞亚群结构的必要步骤,然而目前仍然存在一些挑战。首先,由细胞的自身特征(如细胞所处周期阶段、细胞大小)和技术(捕获方法、捕获效率、PCR扩增、测序深度等)引入的技术噪音和偏差。这些噪音和偏差将导致转录组的基因覆盖极度不均匀,从而造成零覆盖区域和dropouts的产生。另外,当一个队列的多个样本同时进行分析时,样本间的技术偏差和变异将会主导细胞的聚类,导致细胞群体的形成更偏向于不同样本来源而非细胞类型,即批次效应。
在本文中,我们回顾了最近发展的用于提升单细胞转录组聚类效果或其相关的统计和机器学习方法。这些方法涉及:(1)用于基因表达值的标准化、dropouts推测、数据降维以及细胞特异Marker鉴定的数据预处理方法;(2)传统的聚类算法,包括基于划分的聚类、层次聚类、混合模型、基于图的聚类、基于密度的聚类、神经网络、集成聚类和近邻传播聚类等;(3)在时间序列样本和多个批次的细胞群中进行聚类并检测罕见细胞类型的新方法。我们还讨论了单细胞转录组聚类分析中的几个重要方面,包括细胞间相似性度量,特征值提取和单细胞聚类结果的评估。此外,我们对十多个软件包进行了比较,以评估它们在大规模单细胞转录组数据集上的聚类性能和效率。最后,我们对聚类分析中存在的一些挑战进行了讨论。
03
数据的预处理
在单细胞转录组数据的聚类分析中,数据预处理对于减少技术变异和噪声(如捕获效率低、扩增偏差、GC含量、总RNA含量和测序深度的差异等)以及建库和测序过程中产生的dropouts至关重要。高维的基因表达矩阵通常需要经过标准化及降维映射到低维空间中,一些计算方法还利用到统计学和数学方法来解决dropouts事件。
标准化
原始的单细胞转录组数据通常从两个层面进行标准化:细胞的标准化和基因的标准化。细胞的标准化是为了消除扩增偏差和其他细胞特异性的效应,可以通过常用的reads计数标准化方法实现,如FPKM、RPKM、TPM等。基于UMI建库的实验方案,理论上已经避免了与扩增或测序深度相关的误差,因为被相同UMI标记的reads只会统计一次。然而,由于测序文库通常是不饱和的,标准化对于该类型的数据也是有效的。细胞标准化的另一个方法是使用“spike-in”,它的基本思想是,由技术原因带来的误差对于内外源基因的影响是相同的。另外,使用对数转换进行原始计数值的处理也非常常见。
基因标准化的目的是为了防止一些高表达基因主导了分析。常用的基因标准化方法如,在PCA中包含的z-score标准化。从过往的经验中可以看到,基因的标准化可以提高算法的收敛和聚类效果。值得注意的是,数据的标准化处理将会使其失去原本基因表达的相对尺度,并且由于表达值的平移,造成表达矩阵变得不那么稀疏,这可能会影响到大规模数据集的聚类结果。
在SINCERA包中,对基因的标准化方法即是z-score,对细胞的标准化则是使用截尾均值(Trimmed mean)。一些工具会执行更为特殊的标准化。例如,BISCUIT通过学习代表技术误差的参数,在聚类过程中进行迭代标准化;RaceID将每个细胞内的总表达计数标准化到所有细胞表达计数的中位值。
此外,如果基因或者细胞显现出极低的表达信号(基因表达值过低或者细胞表达基因过少),通常会将其移除,因为它们往往代表着虚假信号。在不同的研究中,为去除低表达基因和细胞建立了不同的阈值,这主要根据分析中囊括的细胞和基因的数量而有所不同。例如,scVDMC对PBMC样本的处理中,表达值低于3的基因和总表达计数值小于200的细胞都将被去除。
虽然基因和细胞的标准化在目前大多数的单细胞数据分析流程中是常见的,但关于其对聚类结果的影响仍存在一些争论。一项研究的分析表明,基于bulk的标准化方法在单细胞上的应用可能会对其分析产生严重的不良后果,例如在聚类前进行的高变基因的检测。相同的,也有研究表明,通过中位数或者“spike-in”进行标准化无法解决dropouts存在的问题,反而可能消除每种细胞类型特有的生物随机性,这两者都会导致潜在的细胞类型的不恰当聚类或表征。
通过下面的例子,我们可以认识到标准化的重要性。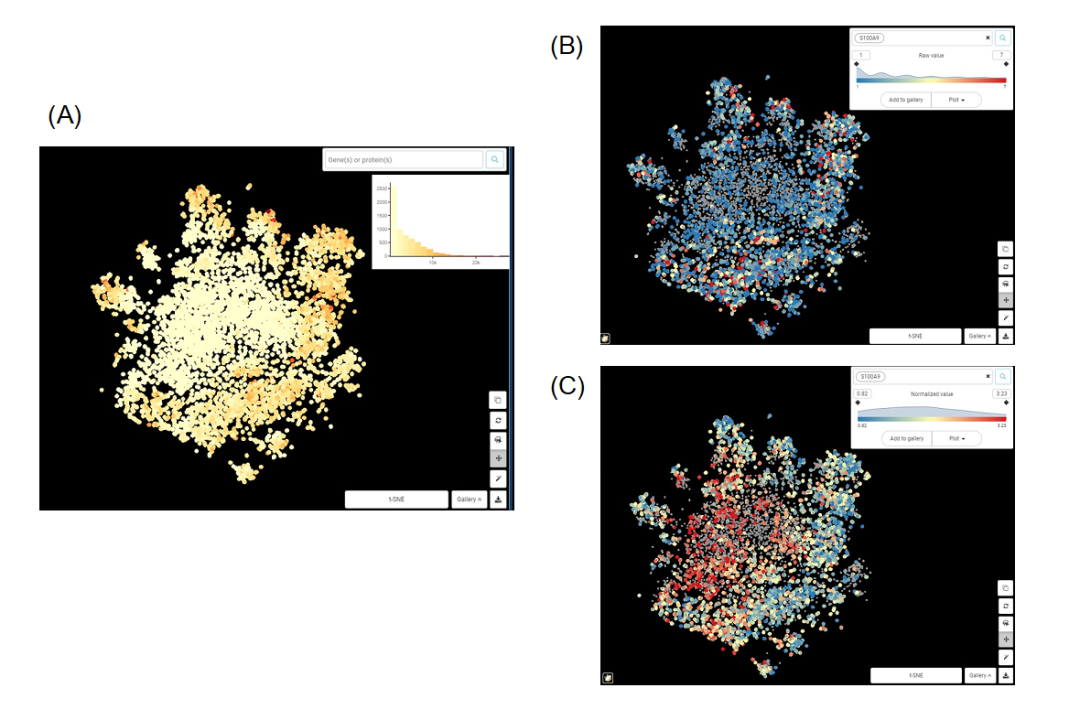
Figure 1. 巨噬细胞群t-SNE图 来自Zilionis等人数据集的巨噬细胞群t-SNE图。(A)依据总计数值上色;(B)依据基因S100A9原始计数值上色;(C)依据标准化后的S100A9的表达值上色。
从图1A,B很容易看出,S100A9的原始表达值与总计数高度相关,两个图的中心区域计数和表达量都较低,而外围区域计数和表达量较高。我们能得出的唯一结论是,当细胞中捕获的转录本总量增加时,S100A9转录本的数量也会增加。这显然没什么意义。而在图1C中,经过标准化后的S100A9表达值与总计数之间似乎没有相关性。我们可以说,S100A9表达的差异不依赖于测序深度等技术噪音,而应该来自(主要)生物因素。
Dropout
单细胞转录组数据中一个重要的技术误差被称为“dropouts”。Dropout事件是指在反转录过程中由于缺失或转录本表达过低而导致基因未表达的错误定量。先前的研究也表明,简单的数据标准化并不能解决该问题。因此,一些聚类算法中包含了特定的机制以矫正dropouts。例如,Seurat通过跨细胞的基因共表达模式,在聚类前进行标记基因的挑选。
另外,也可以通过计算配对相似性来估算dropouts。CIDR便是在聚类前进行缺失值的填补。首先分析单细胞中可能出现的dropouts,识别每个细胞中的候选dropout基因,计算每个基因的dropout率;然后使用候选基因的dropout率来估算表达水平,即当dropout事件以高概率被识别时,检测算法会从其它细胞的表达谱中对该基因的表达值进行填充;最后,利用矫正后的值计算细胞间的不相似度,进行层次聚类。Seurat和SNN-Cliq是基于共享最近邻SNN来度量细胞相似性。已经证明,在稀疏的高维数据中,SNN考虑到周围的近邻数据点,更适合应用于存在dropouts的聚类分析。
在一个更复杂的概率图模型中,BISCUIT明确估计了每个细胞中的基因表达,以及通过数据分布和先验分布估算的代表技术和生物学变异的参数。其中,代表着未观测到的基因真实表达水平的随机变量被引入图模型中并通过吉布斯抽样来估算表达值。
降维
降维通常用于将高维基因表达数据投射到低维空间,使分析聚焦于低维空间中的相关信号,从而更好地实现数据的可视化、聚类分析等,帮助进行生物学解释。当维数大于样本数时,降维还有助于解决样本不足的统计学问题。许多降维方法已经应用于单细胞转录组聚类算法,包括PCA、多维尺度变换(MDS)、t分布、随机近邻嵌入(t-SNE)、典型相关分析(CCA)、潜在狄利克雷分布(LDA)以及嵌入其他模型的降维等等。
PCA:将原本数据点映射到与协方差矩阵的最大特征值相关联的特征向量(即主成分),以保留原始数据中的大部分方差。例如,pcaReduce在聚类前将表达矩阵映射到一个含有K-1个主成分的空间中;SC3使用PCA和拉普拉斯变换应用于距离矩阵以获得一致性矩阵并进行层次聚类。此外,在聚类之后,PCA也被广泛应用于二维或三维的数据可视化。PCA是一种基于假设数据为高斯分布的线性投影方法,为了捕捉数据中的非线性结构,可以使用核主成分分析与非线性核映射相结合。
MDS:也称为主坐标分析(PCoA)。MDS将数据点映射到低维空间,通过最小化所有配对数据点的原始空间中的距离与投影空间中的距离之间的差值,从而在低维嵌入保持原始高维空间中的数据点之间的距离。CIDR便是使用MDS来计算细胞的不相似矩阵。MDS的优点是在低维空间中保持原始的成对距离,易于实现非线性特征嵌入。然而,MDS不能扩展到大规模数据,因为必须计算成对距离来最小化目标函数。
t-SNE:是一种将距离转换为概率的方法。t-SNE构造一个与原始空间及映射后的低维空间中数据点之间的相似性相关的概率分布,然后最小化两个分布之间的Kullback-Leibler散度。t-SNE被广泛应用于单细胞数据分析中的数据可视化。
CCA:是一种基于互协方差矩阵的降维方法。给定两个或多个数据集,该方法查找每个数据集的映射,以最大化数据集之间的相关性。在单细胞转录组的数据分析中,CCA通常用于不同来源样本的整合,如Seurat(图2)。
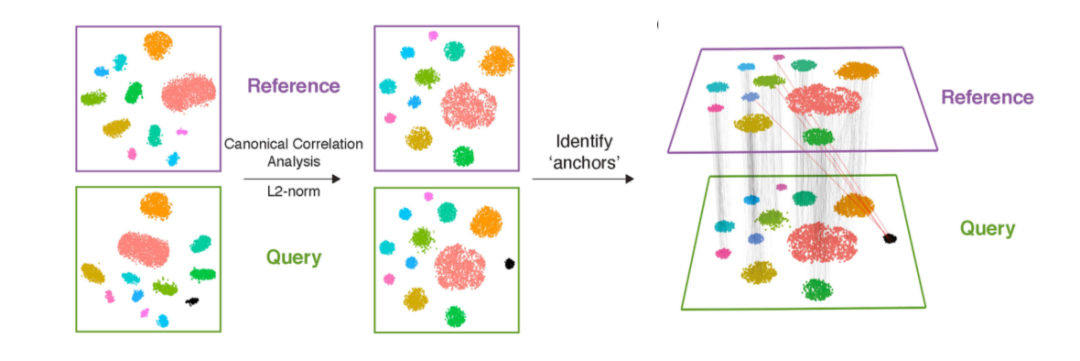
Figure 2. Seurat CCA数据整合示意图
LDA:该方法最初是在自然语言处理中提出的。LDA假设一个文档(document)是通过如下方法生成的:首先从具有狄利克雷先验的话题(topic)的多项分布中对话题进行抽样,然后对文档中的单词(word)进行抽样,这些单词的多项式分布是基于每个话题的狄利克雷先验条件。然后,每个文档都可以在包含k个话题的低维空间中表示。cellTree使用LDA学习“topics”作为潜在特征来表示细胞,其中“words”是受所选的潜在特征制约的基因表达水平。LDA的生成过程产生了一组可解释的潜在特征。
相似度及核函数
在许多聚类方法的计算过程中,不是使用降维的方法,而是通过核函数或相似度函数来计算单个细胞之间的配对相似性进行聚类。核函数策略将从N × M表达矩阵中计算获得N × N相似矩阵,以期望通过核映射或相似函数在隐式特征映射空间中减少原始特征空间中的差异(如果使用有效的核函数)。SNN-cliq和Seurat使用SNN作为相似图。cellTree在用LDA找到的话题直方图上通过卡方找到细胞间的距离。DTWscore利用时间序列样本为每个基因找到细胞对之间的动态时间规整(DTW)距离,以选择高度可变的基因,其中DTW距离是基于两个时间序列在最佳规整路径上的比对计算的。基于TCC的聚类使用细胞间的Jensen-Shannon距离作为谱聚类或近邻传播聚类的输入。SIMLR结合多个核来学习得到细胞相似矩阵,并使用秩约束和图扩散来解决dropouts问题。
大多数其他方法使用更标准的相似性函数或距离函数。BackSPIN,DendroSplit,ICGS和SINCERA在层次聚类策略中使用Pearson相关来寻找最佳分割点。GiniClust和RaceID也分别使用相关性矩阵进行DBSCAN和k-means聚类。参考成分分析(RCA)计算单个细胞和参考细胞之间的表达谱之间的相关性,作为聚类的新特征,以最小化技术差异和批次效应。SC3使用斯皮尔曼、皮尔森和欧氏距离来计算细胞间的配对相似性或距离以获得一致性矩阵。
审核编辑:刘清
-
PCR
+关注
关注
0文章
121浏览量
20484 -
机器学习
+关注
关注
67文章
8561浏览量
137208 -
RNA
+关注
关注
0文章
46浏览量
10118 -
UMI
+关注
关注
0文章
3浏览量
1605
原文标题:单细胞转录组 | 聚类分析中的机器学习与统计方法综述(一)
文章出处:【微信号:SBCNECB,微信公众号:上海生物芯片】欢迎添加关注!文章转载请注明出处。
发布评论请先 登录
微电网暂态稳定分析的前沿方法的适用场景有哪些?

机器学习特征工程:分类变量的数值化处理方法

机器学习和深度学习中需避免的 7 个常见错误与局限性
电能质量在线监测装置支持多维度统计报表吗?
光谱成像技术在作物面积统计中的应用

【新启航】深度学习在玻璃晶圆 TTV 厚度数据智能分析中的应用

XKCON祥控输煤皮带智能机器人巡检系统对监测数据进行挖掘分析
量子机器学习入门:三种数据编码方法对比与应用

统计过程控制在预防性维护中的应用

在ANSA中设置ABAQUS独立非线性分析步的方法

FPGA在机器学习中的具体应用
大模型推理显存和计算量估计方法研究
EM储能网关 ZWS智慧储能云应用(15) — 收益统计




评论