 H掺杂Ga2O3的缺陷计算(热力学稳定性和元素化学势计算TSC)
H掺杂Ga2O3的缺陷计算(热力学稳定性和元素化学势计算TSC)

DASP (Defect and Dopant ab-initio Simulation Package)是一款半导体缺陷和杂质性质的第一性原理计算模拟软件包,该软件包能针对输入的半导体晶体结构,基于材料基因组数据库和第一性原理软件包,自动计算并输出半导体的热力学稳定性,缺陷和杂质形成能及离化能级,半导体样品中缺陷、杂质和载流子浓度及费米能级,关键缺陷和杂质诱导的光致发光谱、载流子辐射和非辐射俘获截面及少子寿命。
针对任一半导体,DASP软件可以计算给出如下性质:热力学稳定性、元素化学势空间的稳定范围、缺陷(含杂质,下同)形成能、缺陷转变能级、各生长条件下的费米能级、载流子和缺陷浓度、缺陷的光致发光谱、缺陷对载流子的俘获截面、辐射和非辐射复合速率等。
本期将给大家介绍DASP HfO2的本征缺陷计算 5.3.2-5.3.2.2 的内容。
5.3.2. 热力学稳定性和元素化学势计算TSC
5.3.2.1. 运行TSC模块
在上一步使用命令dasp 1执行PREPARE模块时,会生成doping-Ga2O3/dec目录,并在该目录中产生1prepare.out文件。等待程序执行完毕,1prepare.out有相应的完成标志。进入doping-Ga2O3/dec目录。确认INCAR-relax,INCAR-static文件中的参数是可行的。(用户可修改INCAR,DASP将根据此目录中的INCAR做后续的计算)
确认PREPARE模块完成后,回到doping-Ga2O3目录,使用命令dasp 2执行TSC模块。同样地,TSC模块会在doping-Ga2O3目录中生成名为tsc的目录,里面记录了TSC程序的计算输出,包括各计算目录以及运行日志文件2tsc.out。等待程序完成期间无需额外操作。
5.3.2.2. TSC模块运行流程
host结构的总能计算(与MP参数保持一致):
TSC模块将使用与MaterialsProject数据库完全一致的输入参数(INCAR,KPOINTS,POTCAR)来对用户给定的原胞做结构优化和静态计算。因此,该计算得到的总能与MP数据库的总能是可比的。此步骤是为了得到影响Ga2O3稳定性的关键杂相。通过目录可以看到:
从doping-Ga2O3/tsc/2tsc.out中也可以看到程序的运行日志,即产生输入文件、relaxation1、relaxation2、static、数据提取等步骤。
关键杂相判断:
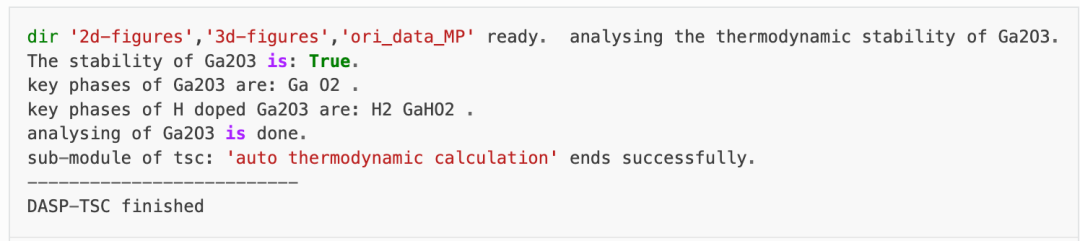
TSC模块将搜寻MP数据库上所有与Ga2O3相竞争的杂项,通过DFT计算的Ga2O3的总能与MP数据库中杂相的总能,判断出Ga2O3是稳定的。
随后,程序将自动下载影响掺杂H的Ga2O3稳定性最关键的杂相,本例中为H和GaHO2。在2tsc.out中可看到相关的信息:

host与杂相结构的总能计算(PREPARE模块确定的参数):
在确定关键杂相后,TSC模块将使用PREPARE模块确定的参数(AEXX)计算Ga2O3,GaHO2,H2的总能。2tsc.out如下:

化学势的计算:
根据DFT计算的总能,计算掺杂H的Ga2O3的形成能和化学势稳定区间。由于Ga2O3是二元的,TSC模块给出2个化学势的端点值,即Ga-rich和O-rich,写入dasp.in:

在2tsc.out可以看到程序执行完毕的输出:
对于三元以上的体系,TSC模块将输出稳定区域图像,及稳定区域各端点处的化学势。
审核编辑 :李倩
-
模块
+关注
关注
7文章
2822浏览量
52798 -
热力学
+关注
关注
0文章
45浏览量
9506 -
软件包
+关注
关注
0文章
113浏览量
12089
原文标题:产品教程丨H掺杂Ga2O3的缺陷计算(热力学稳定性和元素化学势计算TSC)
文章出处:【微信号:hzwtech,微信公众号:鸿之微】欢迎添加关注!文章转载请注明出处。
发布评论请先 登录
《炬丰科技-半导体工艺》Ga-N 系统的热力学分析
聚合物溶液热力学模型的评述
什么叫热力学温度_热力学温度与摄氏温度的关系详解
H掺杂Ga2O3的缺陷计算(准备计算PREPARE02)

H掺杂Ga2O3的缺陷计算(准备计算PREPARE01)
H掺杂Ga2O3的缺陷计算(缺陷形成能和转变能级计算DEC)


双钙钛矿材料稳定性的快速计算预测(Cs2AgBiCl6 01)



双钙钛矿材料稳定性的快速计算预测

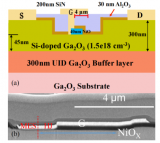
氧化镓(Ga2O3)沟槽二极管的相关研究进展
热力学稳定的双改性LiF和FeF3层赋予高镍阴极卓越的可循环性





 工商网监
工商网监
评论