 基因组重测序的应用领域有哪些
基因组重测序的应用领域有哪些

背景介绍
1.1
基因测序在癌症领域上的应用
癌症是目前人类所面临的最大敌人,其发病率随着年龄增长而升高,在人口老龄化加剧的当下,全社会的癌症负担也将愈发严峻。癌症难以治愈的原因之一是肿瘤具有强异质性,即相同症状、相同病理改变却可能由完全不同的基因变化而造成,以至于同类型癌症患者对于相同药物的药效反应有很大的差别 。这些基因上的变化包含了单碱基突变(SNV)、小片段插入缺失(InDel)、结构与拷贝数变异(SV & CNV)和甲基化等。随着近几年基因测序成本如图 1所示不断下降,在万元内即可完成人类的全基因组测序,GPU的技术发展也带来分析成本与时间的下降,于是用于检测基因组变化的重测序技术在癌症治疗中起到了越来越重要的作用。基因组重测序的应用主要有三个方向:
01
基因测序技术提供肿瘤内基因分子水平的变化,为研究提供预见性,支持肿瘤新靶向药物研发。
02
提高肿瘤早期诊断准确率,降低高危风险,从而提高癌症存活率。以在儿童中高发的儿童神经母细胞瘤(NB)为例,目前国内低、中危组NB患儿,长期无瘤生存率可达90%,但高危组NB患儿的5年总生存率不足30%。而如果能够做到早期诊断的话就可以通过手术切除联合化疗的方式进行治疗,从而提高治愈率。
03
基于易感基因分子水平的基因变化检测,可以根据变化情况为患者提供个体化和预见性的治疗。
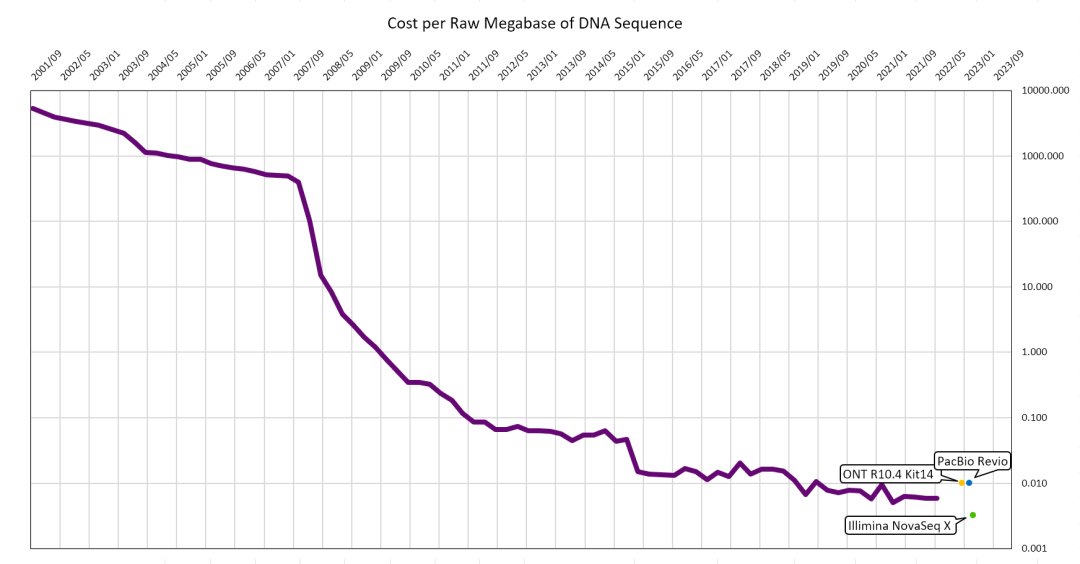
图 1 每兆数据DNA测序开销
图中数据来源于Wetterstrand KA. DNA Sequencing Costs: Data from the NHGRI Genome Sequencing Program (GSP) 和 Next-Generation-Sequencing.v1.10.25
1.2
基因测序手段介绍
当下基因组重测序常见的测序手段分为以Illumina双端150bp为代表的短读长测序与以PacBio HiFi或Oxford Nanopore Technology(ONT)为代表的长读长测序两种。如图1所示,Illumina双端测序将DNA打断两端加上不同接头与流动池中分布的接头结合不断合成、退火与扩增,扩增后利用带荧光的dNTP与流动池中的DNA链结合,由于不同的dNTP发出不同荧光,于是可以通过扫描流动池中的荧光信号,得到序列信息;PacBio 将单分子与DNA聚合酶固定在零模波导孔上,激光从孔的底部照入,激光不能穿过小孔且只能照亮孔底的一小部分区域。当DNA聚合酶使用游离的dNTP进行聚合反应时,dNTP上的荧光基团被激光激发,通过传感器将光信号转化为电信号。ONT则是通过让 DNA 单分子通过带电的纳米孔,不同的碱基使得电流发生改变,传感器记录电流变化,测序仪将电流变化转化为碱基值。
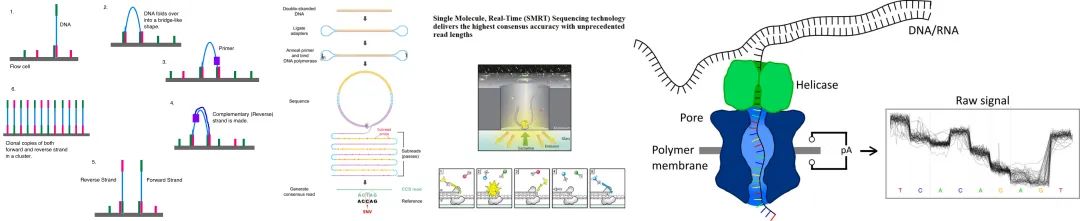
图 2 测序原理示意图 (a) Illumina 双端测序,
图片来源于DMLapato, Illumina dye sequencing, https://en.wikipedia.org/w/index.php?title=Illumina_dye_sequencing&oldid=1159305769#cite_note-Illumina,_Inc_2013-7
(b) PacBio 零模波导孔单分子实时测序,
图片来源于百迈客生物, pacbio三代测序仪, http://www.biomarker.com.cn/technology-services/pacbio
(c) ONT 纳米孔测序,
图片来源于 Kraft, Long-read sequencing in human genetics, DOI: 10.1007/s11825-019-0249-z
短读长测序的优势在于价格低廉与测序碱基准确度高,长读长测序拥有以下几个优点:
01
长读长测序使用单分子测序,不需要扩增反应。一方面节省了时间,另一方面也避免了扩增中引入的错误,比如扩增效率不同带来的偏好性问题。
02
长读长测序相比于短读长测序更容易跨过基因组的重复区域。在短读长测序结果中,至少748个与人类相关的基因存在测序暗黑区域,这个区域序列的Read会出现低深度、低覆盖度及低比对质量。而造成这个现象的主要原因是在参考基因组中该区域的序列多次重复。而测序序列长到能够跨过重复区域的长读长测序正好解决了这个问题。
03
长读长测序相比于短序列可以更容易得到核酸的甲基化水平。短读长测序基于荧光信号,通常需要将样本分为两份,一份利用亚硫酸氢盐处理将未甲基化的C转化为U,另一份直接测序进行比较获得基因组甲基化位点。而基于电信号的三代测序可以直接区分出甲基化的C与未甲基化的C,避免了亚硫酸氢盐等药品处理和多次测序引入的错误。
而近几年来测序的技术有两个明显的发展趋势:
01
短读长测序和长读长测序成本均逐步降低。短读长测序成本降低幅度变缓,长读长测序的价格随着仪器的升级已经降到了三年前短读长测序成本。在一些应用场景中,比如结构变异的检测,由于长读长测序只需要三分之一短读长测序的数据量,长读长测序的成本已经低于短读长测序。
02
随着试剂与算法的研发,长读长测序的准确度也逐渐接近短读长测序。在单碱基质量和测序价格的保证下,长序列能保留更原始的基因组特征。
于是目前研究所选择的测序手段逐渐从短读长测序向长读长测序转移。
1.3
基因组重测序的流程介绍
如图3所示,重测序的流程主要包含以下大步骤:
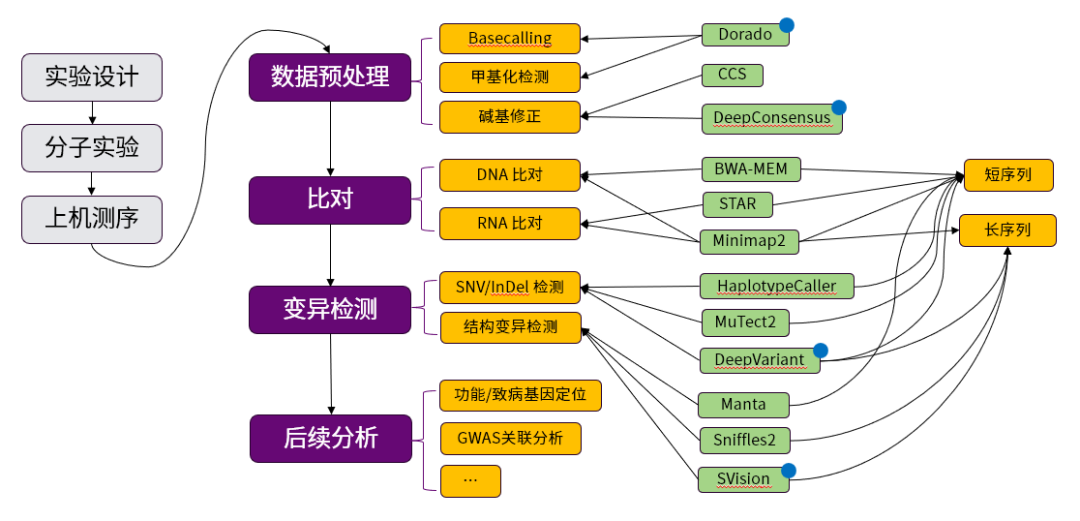
图 3 基因组重测序流程与软件示意图。
图中紫色矩形部分为主要分析步骤,黄色矩形部分为分析类别,绿色矩形部分为软件,蓝色上标的软件为利用到了机器学习的软件。
01
实验设计、分子实验与上机测序:这一步包括了需求分析和选择测序手段。分子实验通常包括核酸提取、核酸分子打断和样品的制备。上机测序将样品加入测序仪中,测序仪产生序列信息,完成测序。
02
数据预处理:通常测序仪会利用传感器捕获电信号,测序仪的Basecalling步骤和甲基化检测步骤将电信号转化为文本形式的序列。基因组测序的结果保存检测得到每一个核酸分子的碱基和该碱基的质量值,ONT测序和PacBio测序也会保存原始电信号。甲基化检测的结果会同时输出每一个碱基的甲基化概率。长序列检测会发生随机的测序错误,被认为准确度不高,需要进行碱基修正,修正测序错误来提升碱基质量值。
03
比对:这一步骤将测序得到的序列与参考基因组比对,确定该序列来自于参考基因组的哪个部分。RNA测序得到的序列相比于DNA缺失了内含子部分,长读长测序与短读长测序在错误率、复杂度上有区别。所以软件上会根据样本的类型和测序序列长度做区分。
04
变异检测:变异检测将获得样本与参考基因组的差异,这些差异包括了单碱基的变异(SNV)、小的插入缺失(InDel)以及大的结构变异(SV)。短读长测序由于单碱基的准确度高,在SNV和InDel上有优势,而对于长度大于短读长测序序列长度的结构变异而言,长读长测序更有优势。所以软件上会根据需要检测的变异长度和测序序列长度做区分。
05
后续分析:后续分析则包括了变异的过滤、注释、比较以及关联分析等过程,这一步将以万计的变异中筛选出与研究课题息息相关的变异,会跟随着课题不同而不同。
而这五个大步骤之中,第一步的时间改进则依赖于实验设计的改良以及试剂的研发。而后续步骤则需要依赖于算法的改进和计算设备的提升,在本文中将会介绍这些分析的常用软件和算法,并指出这些软件均可以通过对GPU和并行算法的优化进行提速。比如Carpi等人利用合理实验设计和GPU,使得恶性疟原虫在计算成本减少4.6倍的同时整体分析速度提升27倍。
二
测序数据预处理
2.1
利用GPU获得碱基序列
将测序过程中产生的电信号转化为碱基的步骤被称为 Basecalling。以 ONT测序为例,如图 4所示,当核酸分子通过芯片上纳米孔时,生成不断变化的电信号,测序仪记录下电信号,并转化为碱基。
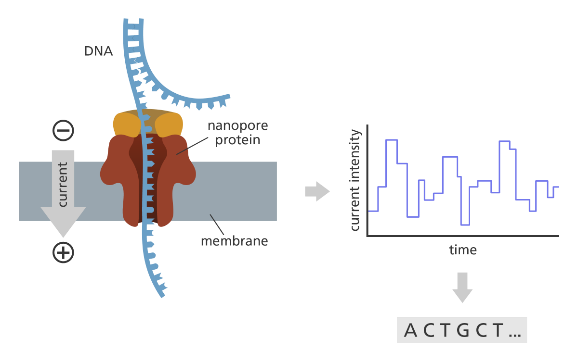
图 4 ONT Basecalling 过程示意图
图片来源于What is Oxford Nanopore Technology (ONT)sequencing?,https://www.yourgenome.org/facts/what-is-oxford-nanopore-technology-ont-sequencing/
由于碱基通过纳米孔的速度并不匀速,电流信号与预测得到的碱基单调但非对齐,为了解决这个问题,ONT判断碱基的序列则是通过语音识别中常用的 CTC 作为损失函数来实现。ONT开源Basecalling 软件Dorado 的r10.4.1系列模型中,模型分为编码器和解码器两个部分,解码器的模型如图 3所示,其接续线性条件随机场(CRF);解码器则是用Viterbi算法进行的CRF解码。Dorado对每一个测序仪器构建Fast、Hac 和 Sup 三种模型,模型依次增大且得到的碱基结果逐渐准确。r10.4.1模型中,在Fast 模式下,解码器进入到线性CRF层的特征向量为96,状态长度为3;Hac模式为特征向量128,状态长度为4,而Sup模式特征向量达到了256,状态长度为5。在 Fast模型下GPU加速效果可以达到60倍以上。

图 5 Dorado r10.4.1 Fast模式的模型示意图
2.2
DeepConsensus深度学习修正测序错误
由于PacBio会有完全随机的错误发生(主要为短片段的插入与缺失),PacBio使用同一个零模波导孔内SMRT 圈多次测序得到多条Subreads序列的方法去除测序错误,矫正得到一个低错误率高质量值的序列,平均SMRT 圈的测序圈数决定了测序的时间。PacBio Revio测序仪使用CCS 和DeepConsensus 模型来矫正测序错误获得高质量的序列。CCS基于隐马尔可夫模型,其GPU版本可加速至12倍。DeepConsensus是一个编码器-only的Transformer模型,与使用CCS的结果相比,DeepConsensus能够得到更准确的多聚核苷酸位点。
DeepConsensus将测序得到的Reads切割,每份 100bp,每一份向量包含了序列、链方向、脉冲宽度、脉冲间隔以及信噪比等信息。通过一个6个自注意力层的编码器后,使用以Softmax作为激活函数的前馈神经网络解码得到真实的序列。由于GPU在神经网络训练和推理上的优势,相比于CPU版本的DeepConsensus,GPU加速效果能够达到 3.3倍以上。
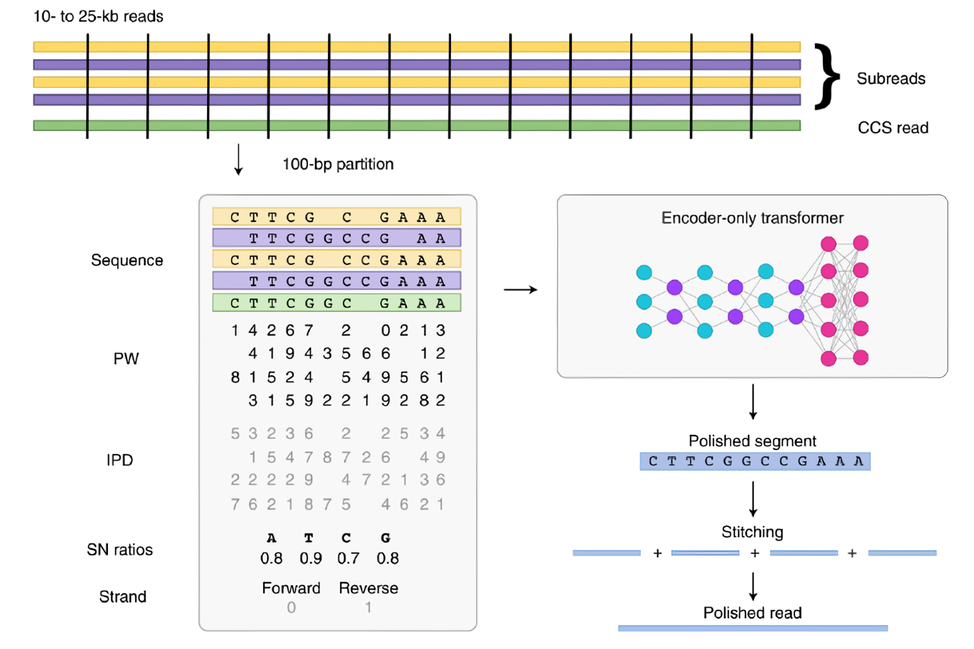
图 6 DeepConsensus 流程示意图
图片来源于Baid et al. DeepConsensus improves the accuracy of sequences with a gap-aware sequence transformer. DOI: 10.1038/s41587-022-01435-7
三
测序序列比对
两条序列比对问题是序列比对的基础,以图7所示,序列1 GACTTTCC 与 序列2 TAGACTACC 进行比对得到比对表。两条DNA序列的比对通常有四种状态:插入、缺失、匹配和错配,而匹配和错配往往会在比对问题中合并为同一个状态。

图 7 seq1与seq2的序列比对示意图
Smith-Waterman算法和 Needleman-Wunsch算法是序列比对中常用的算法,其中Smith-Waterman算法用于局部比对而Needleman-Wunsch算法应用于全局序列比对。而实际案例中,由于参考基因组的序列长,比如人的参考序列总长度在3Gb,一次测序深度10x测序所得Reads数目在2百万左右,直接使用局部比对的Smith-Waterman算法在时间成本上是不可接受的,于是有两种加速方案:第一种为各测序所得序列间并行运算,另一种则是对现有算法进行改进,Minimap2等基Needleman-Wunsch的比对软件便应运而生。在本节中首先介绍Smith-Waterman与Needleman-Wunsch的动态规划算法与其GPU加速情况,然后介绍Minimap2特殊的GPU加速。
3.1
Smith-Waterman与Needleman-Wunsch 算法与GPU加速
两个算法拥有相同的步骤:首先初始化得分矩阵,根据相似性矩阵和罚分矩阵填满表格,后根据分数回溯得到比对结果。如图 8所示,Gotoh提出的比对方式具体如下:
01
确定罚分矩阵,罚分矩阵定义了碱基匹配的奖励分s、开启间隔区域(插入或者缺失)的惩罚分q和持续间隔e的惩罚分。这一步往往根据测序的特性进行确定。
02
根据罚分矩阵来初始化打分矩阵,通常会将分数存储在三个打分矩阵 H、E和F中,分别代表着匹配分数、插入分数和缺失分数。Smith-Waterman算法中,打分矩阵H、E 和F的首列与首行初始化为0;而Needleman-Wunsch算法中,打分矩阵F首列初始化公式为F0,j=F0,j-1-e,打分矩阵E首行初始化公式为Ei,0=Ei-1,0-e,打分矩阵H的首列和首行初始化为Hi,j=max{Ei,j,Fi,j}
03
计算打分矩阵H算法中的每一个位置的分数Hi,j 。Smith-Waterman 算法中Hi,j=max{Hi-1,j-1+s,Ei,j,Fi,j,0} ,而Needleman-Wunsch算法中,Hi,j=max{Hi-1,j-1+s,Ei,j,Fi,j},其中 Ei,j=max{Hi-1,j-q,Ei-1,j}-e,Fi,j=max{Hi,j-1-q,Fi,j-1}-e。
当打分矩阵H、E和F全部填写完成后,需要回溯来获取最优的比对序列。Smith-Waterman算法从打分矩阵中的最大值开始回溯直到Hi,j=0为止;而Needleman-Wunsch算法从打分矩阵H右下角开始回溯,直到左上角为止。回溯路径即为比对路径。此时对于长度M和长度N的序列对而言,时间复杂度为O(MN)。
Myers提出了Bit-Vector加速该算法,打分矩阵Hi,j中的依赖于Hi-1,j-1、Hi-1,j和Hi,j-1,如果以列向量的角度看的话,如图 9所示,那么Hj仅依赖于Hj-1。该算法将计算打分矩阵Hi,j变为计算行差值Δhi,j与列差值Δvi,j。将时间复杂度降为O(N)。
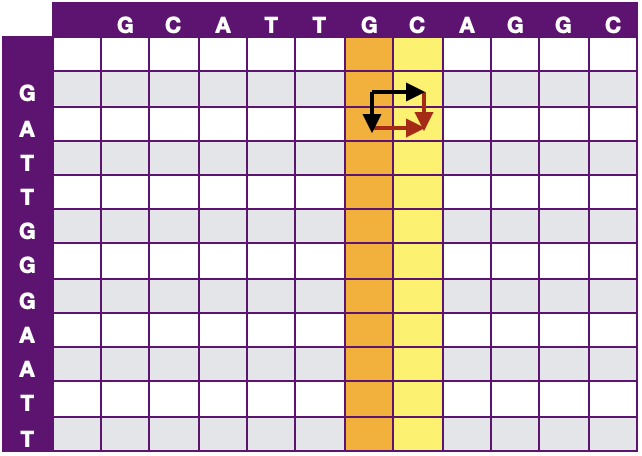
图 9 Bit-Vector 加速示意图
Siriwardena等人提出了另一个加速方案,如图 10所示,次对角线上的各个元素没有相互依赖,故拓展到次对角线的Block没有依赖,因此通过各个Block之间的并行计算进行GPU加速。该方案运行时间与比对序列长度成非线性正相关,在如人染色体的数量级上能够达到40倍的加速效果。
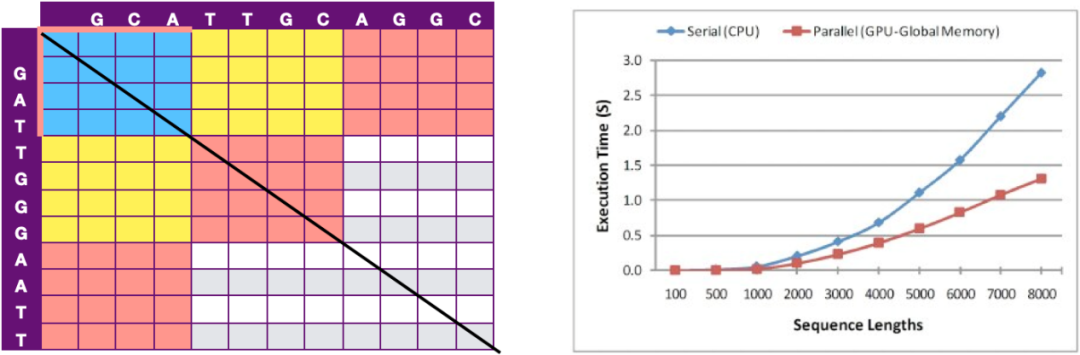
图 10 次对角线分块加速示意图
与CPU 和 GPU执行时间比较图
除了上述提到的方案以外,使用动态规划算法的时候需要频繁用到max、min等计算,GPU同时采用特殊的硬件指令来加速动态规划算法,预计可以加速7倍。
3.2
比对软件的新星Minimap2的流程与GPU加速
为了解决Smith-Waterman局部比对在大规模数据中耗时长的问题,Li开发了Minimap2 软件极大减少了比对的时间。Minimap2 是在比对领域较新且广泛使用的软件,相比于其它软件如BWA-mem、STAR等有特定使用场景的限制,Minimap2的使用范围比较广泛,其适用于二代的DNA短读长测序数据以及三代DNA长读长测序数据,也可以被应用RNA-seq这种有大间隔的数据。如图11所示,Minimap2将测序得到的 Reads与参考基因组的比对的过程主要分为 Seeding、Chaining 和 Alignment 三个步骤。
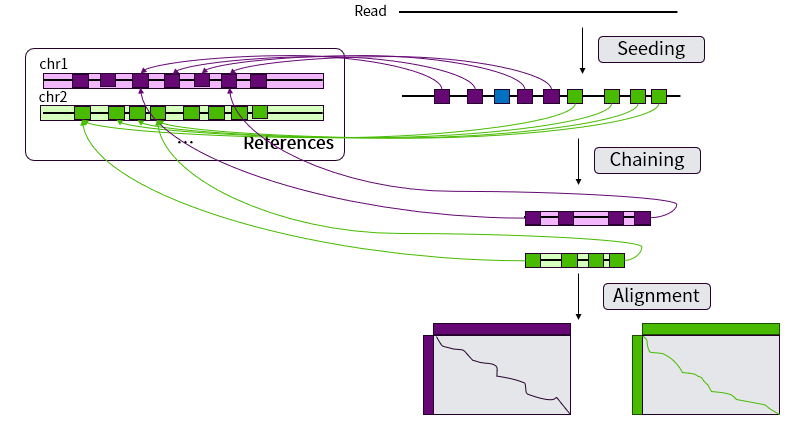
图 11 Minimap2 流程示意图
01
Seeding这一步快速定位参考基因组与Read之间完全匹配,无插入与缺失的短序列,得到若干有匹配位置信息的锚点(Anchors)。锚点使用A(x,y,w)表示,意味着参考基因组的坐标[x-w+1, x]与Read的坐标[y-w+1, y]完全匹配,每一条Read会得到若干个锚点。
02
Chaining这一步则是将Seeding 这步得到的锚点连起来,确定这条Read在参考基因组上的真实位置。在 Minimap2中,Chaining是用一个一维的动态规划算法计算f 解决的,将所有锚点根据参考基因组的位置坐标x进行排序,第 i个anchor的分数f(i)可以由公式 f(i)=max{max{f(j)+α(j,i)-β(j,i),wi}(i>j≥1)}, 计算得到,其中 α(j,i)=min{min{yi-yj,xi-xj},wi} 作为奖励分。为两个锚点之间相同的长度;β(j,i)=γc ((yi-yj )-(xi-xj )),作为两个锚点之间发生插入或者缺失间隔的罚分,γc 是一个与间隔长度相关的函数,可以调整 γc 以适应不同测序类型。Minimap2也会设置一个最长间隔G,当max{yi-yj,xi-xj}>G时,罚分β(j,i)=∞。若有N个锚点,那么直接计算最大f时间复杂度为O(N2),基于大部分f(i) 的最大值在i附近,故只会计算h次,于是Minimap2将时间控制在O(hN)内。对于i每次得到f后,会回溯到 j 直到f(j)=wj ,此时将回溯过程中的锚点记为“已用”,得到一条链,每一个链根据匹配长度和间隔长度计算比对分数S。当我们将所有的锚点标记为“已用”后,可以得到所有链和其分数S,分数最高的链便是得到的最优链。
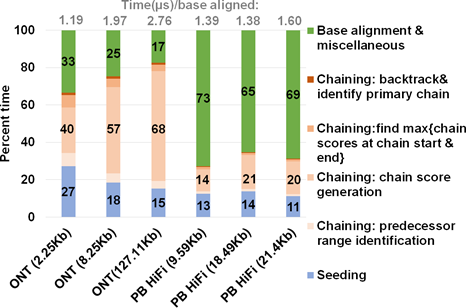
图 12 Minimap2不同步骤所花时间比例图。图片来源于Sadasivan et al. Accelerating Minimap2 for Accurate Long Read Alignment on GPUs. DOI: 10.26502/jbb.2642-
03
Alignment 这一步,则是将测序得到的 Read 与 Chaining 这一步得到的链序列进行 base to base 的全局比对。这一步则可以用上一小节所提到的Needleman-Wunsch算法完成。
Minimap2的耗时时间如图 12所示,Chaining和 Alignment 是耗时最长的时间。因此提升 Chaining 和 Alignment步骤的速度是提升 Miniamp2 运行速度的关键。
Sadasivan 等人提出了Minimap2 Chaining步骤的并行方案。如图 13所示,他们首先将 Minimap2 的Chaining步骤的逆向搜索变成顺向搜索,并且对于每一个i锚点并行计算其顺向的 >i 锚点是否为最大分数。GPU通过这种方式并行即可以将Minimap2提速3.8倍左右。
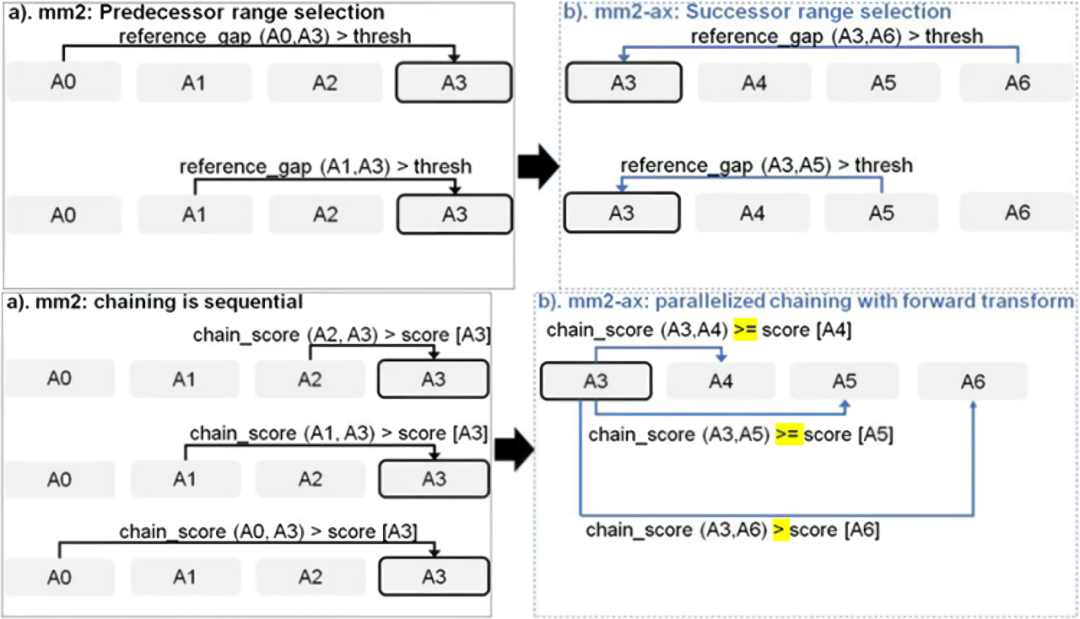
图 13 mm2-ax 加速 Minimap2 Chaining 的方案
图片来源于Sadasivan et al. Accelerating Minimap2 for Accurate Long Read Alignment on GPUs. DOI: 10.26502/jbb.2642-91280067
四
变异检测
4.1
SNV与InDel的检测
在生物学上,通常需要关注的是测序得到的变异。单碱基变异(SNV)是常见的一种变异方式,SNV 是单碱基的变异,碱基的突变有时候会改变氨基酸,或者使得转录提前终止。SNV的检测主要分为两个步骤:1. 确定并过滤变异位置; 2. 确定基因型。在本节中将介绍两种软件,一种是以 HaplotypeCaller和MuTect2为代表的基于传统概率学模型建模的软件,另一种则是以DeepVariant为代表的利用深度学习进行检测的软件。
4.1.1
概率模型 HaplotypeCaller / MuTect2检测流程
HaplotypeCaller 与 MuTect2 是 GATK 中重要的 SNV 与 InDel Calling 的软件。两款软件在一定程度上拥有相同的原理,在概率模型上的区别,导致HaplotypeCaller 主要用于性细胞变异的检测,而MuTect2 更适用于体细胞变异的检测。如图 14两款软件都在确定变异区间后,利用局部组装得到的单倍型并将测序得到的Reads用Smith-Waterman算法比对到单倍型中来减少测序、比对等错误信息,通过 PairHMM 来得到每一个单倍型的似然值,然后根据各自的概率模型获得最终的基因型。
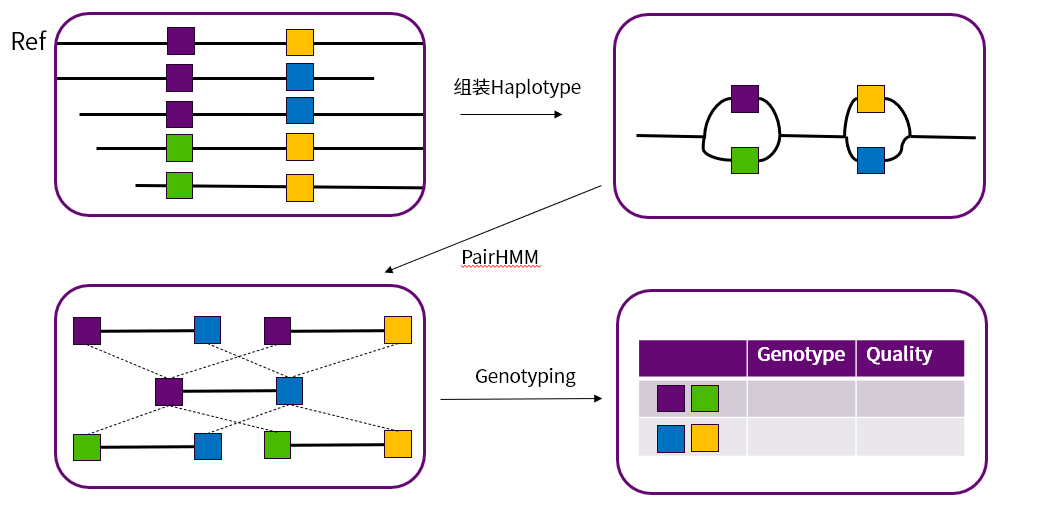
图 14 HaplotypeCaller与MuTect2 的通用流程示意图
每一个单倍型的似然值时HaplotypeCaller和MuTect2的关键步骤,即求单倍型的集合H对于所有测序得到的Reads的集合R的比对结果集合A的概率,即 P(R│H)=∑AP(R,A|H),那么只需要求解每一个比对的概率即可。其中我们已经看到了重新比对中Smith-Waterman的GPU加速方案,HaplotypeCaller中的PairHMM这一步亦可以用GPU并行加速。
PairHMM 的过程与前述的比对算法相似,定义了三个状态: 匹配M, 插入I和缺失D。如图15所示,三个状态会发生转移, M→I或者 M→D,即从匹配状态转化为插入状态或者缺失状态概率为δ、 I→M 从插入状态结束间隔变回匹配状态概率为ε、D→M从缺失状态结束间隔变回匹配状态概率为ε,也会发生持续 M→M 即延续匹配状态概率为1-2δ、I→I 延续插入状态概率为1-ε和D→D 延续缺失状态,概率为1-ε。基于生物的序列特征,往往1-ε<δ≪1-2δ,即维持间隔的概率小于发生间隔的概率,远小于匹配的概率。
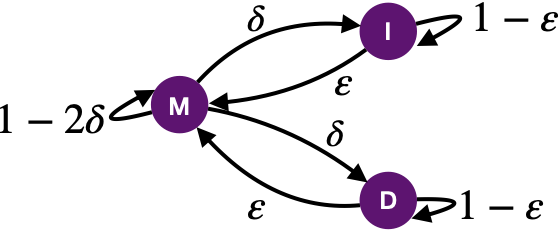
图 15 PairHMM 状态转化示意图
计算时定义了三个状态分别定义了一个矩阵 M、I和D。Mi,j表示测序序列i与参考基因组j位置匹配的概率,同理 Ii,j和Di,j分别代表着发生插入的概率和缺失的概率。矩阵M可以通过公式 Mi,j=S(Mi-1,j-1 (1-2δ)+Ii-1,j ε+Di,j-1 ε) 计算而得,其中S与碱基的质量值错配概率等有关;矩阵I可以通过公式Ii,j=Mi-1,j δ+Ii-1,j (1-ε) 得到;矩阵D可以通过公式Di,j=Mi,j-1 δ+Di,j-1 (1-ε) 得到;当计算完矩阵M、I后,将M与I的最后一列相加即可得到似然值。
PairHMM是检测流程中最密集的部分,因此提升 PairHMM的速度就是提升 HaplotypeCaller/MuTect2 的关键,可以发现PairHMM的分数矩阵计算过程与Smith-Waterman的打分矩阵过程相似,于是Li 等人参考Smith-Waterman的GPU加速方案提出的基于 GPU的 PairHMM 比CPU版本加速 783 倍。
4.1.2
DeepVariant深度学习检测SNV
HaplotypeCaller/Mutect2 传统的概率模型算法存在两个问题:
1
由于不同的测序技术或者物种不同,HaplotypeCaller所需要的往往先验参数不同。比如PacBio测序中,插入发生的概率大于缺失发生的概率,远大于传统的 Illumina 二代的概率,于是 HaplotypeCaller 需要根据测序技术的更迭不断尝试获得最优的先验参数。
2
传统概率模型的建模中假设了SNV在基因组发生的概率相同。而由于自然选择的存在,SNV的发生概率在基因组中并非均匀。在生物中,往往位于内含子区域的变异发生概率会大于外显子区域概率,而管家基因内变异发生概率会远小于其它基因变异发生概率。
为了解决这个问题,DeepVariant与 HaplotypeCaller 不同,利用深度学习的方法代替传统的概率模型。如图 16所示,在确定变异的位置后,DeepVariant将覆盖该位置的 Reads 提取出来,组成一个 PileUp 的图像,图像包含了该位点上下游100bp的序列、质量值、Reads 链、比对质量、变异支持及覆盖情况等信息,这些信息将起到与 HaplotypeCaller 组装单倍型的相似作用。

图 16 DeepVariant MakeExample 所以生成的典型图片,分别为纯合SNV变异、杂合SNV变异和纯合未变异。图片来源于Looking Through DeepVariant’s Eyes. https://google.github.io/deepvariant/posts/2020-02-20-looking-through-deepvariants-eyes/
得到图片后,DeepVariant 使用 CNN 将 PileUp 得到的图片推理直接获得该位点基因型的似然值,从而判断是否发生变异以及变异的基因型。
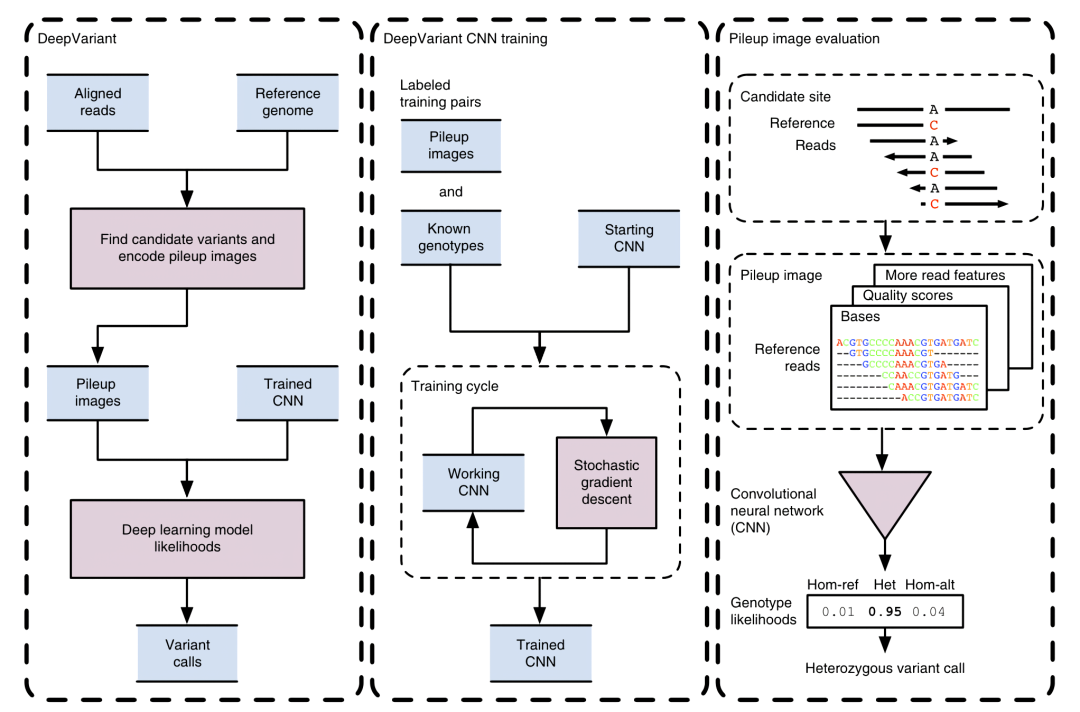
图 17 DeepVariant流程示意图
图片来源于Poplin et al. A universal SNP and small-indel variant caller using deep neural networks. DOI: 10.1038/nbt.4235
DeepVariant 在测序技术和物种都具有泛化性,用户可以通过两个个体的测序数据构建纯合和杂合的“银标准”对DeepVariant的模型进行微调,使DeepVariant适合不同的测序技术(如 Illumina、PacBio HiFi、 ONT R10.4.1等等)。另一方面,DeepVariant 也通过对输入或者输出改造,已经衍生出应用于其他场景的模型,比如适用于家系的 DeepTrio,或者利用肿瘤样本-正常细胞样本对检测体细胞变异的 VarNet。如图 18所示,DeepVariant 在 GPU 的帮助下可以显著减少 Variants Calling 这个步骤。
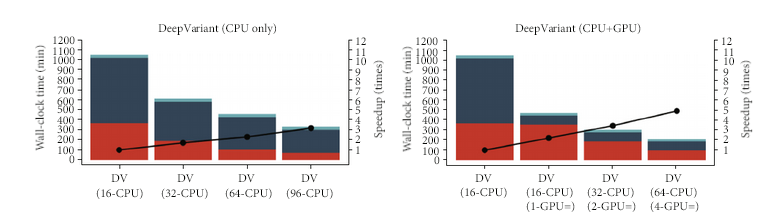
图 18 DeepVariant不同硬件不同环节时间统计图。
图片来源于 Huang et al. DeepVariant-on-Spark: Small-Scale Genome Analysis Using a Cloud-Based Computing Framework. DOI: 10.1155/2020/7231205
4.2
结构变异检测
结构变异通常包括了插入、删除、串联重复、染色体倒位、染色体易位、拷贝数变异和复杂的复合结构变异等变异。结构变异涉及到>50bp的序列,与SNV或小片段的InDel相比,一旦变异发生通常更不可逆。结构变异的检测有着重要的意义:
1
由于不可逆的特性,结构变异往往是人种区别、物种分化的关键。
2
稀有的结构变异往往与疾病或者癌症发生关联。
3
在肿瘤细胞中变异会出现大量的高密度的异常的结构变异,甚至会出现染色体碎裂的现象。
结构变异的检测的方法如图 19所示,包括了基于双端测序方向的Read Pair、基于测序深度变化的 Read Depth、基于比对时异常比对的Split Reads 和将测序得到的 Reads组装与参考基因组比对的 Assembly。目前短序列有Manta、Delly,而长序列有 Sniffles2、CuteSV等软件,基于比对时异常比对的Split Reads是主流的方法。
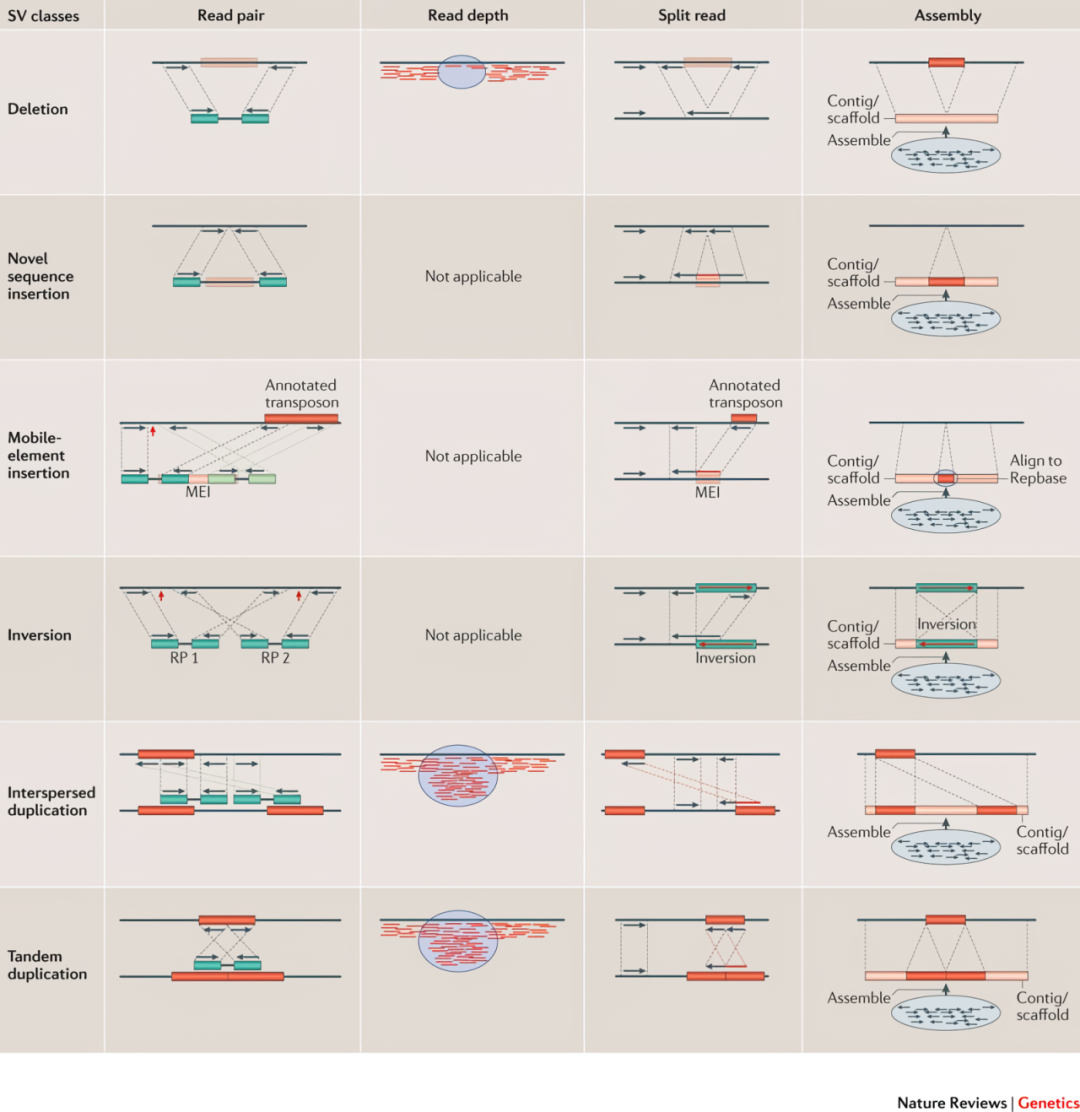
图 19 结构变异检测的常用方法
图片来源于Alkan et al. Genome structural variation discovery and genotyping. DOI: 10.1038/nrg2958
4.2.1
SVision深度学习模型
如 Sniffles2、Manta等结构变异软件,过滤得到比对异常的Reads,将比对异常点根据其在参考基因组的位置、异常的长度等方式进行聚类,每一个类即获得一个结构变异位点。但是这缺少了发现复合结构变异的能力。通常复合结构变异检测都依赖于现有的模式,如易位后缺失等,缺乏一个统一的结构。
SVision 便是为了解决这个问题而出现的模型31。如图 20所示,SVision包含了编码器、tMOR和解释器。编码器将测序的序列转化为图片,其识别支持变异序列和参考基因组序列的碱基通过VAR-REF和REF-REF图像,从VAR-REF中去除REF-REF中已有的重复序列,以提高后续分析的准确度。在tMOR步骤中,SVison将包含多个复合结构变异断点的图像通过5层卷积层、1层展平层和2层全连接层的卷积神经网络进行预测发生变异的概率。最后,解释器描述了不同的复合结构变异。由于这是一个卷积神经网络的模型,GPU的矩阵乘法上的优势能够比CPU版本的SVision快1.8倍。
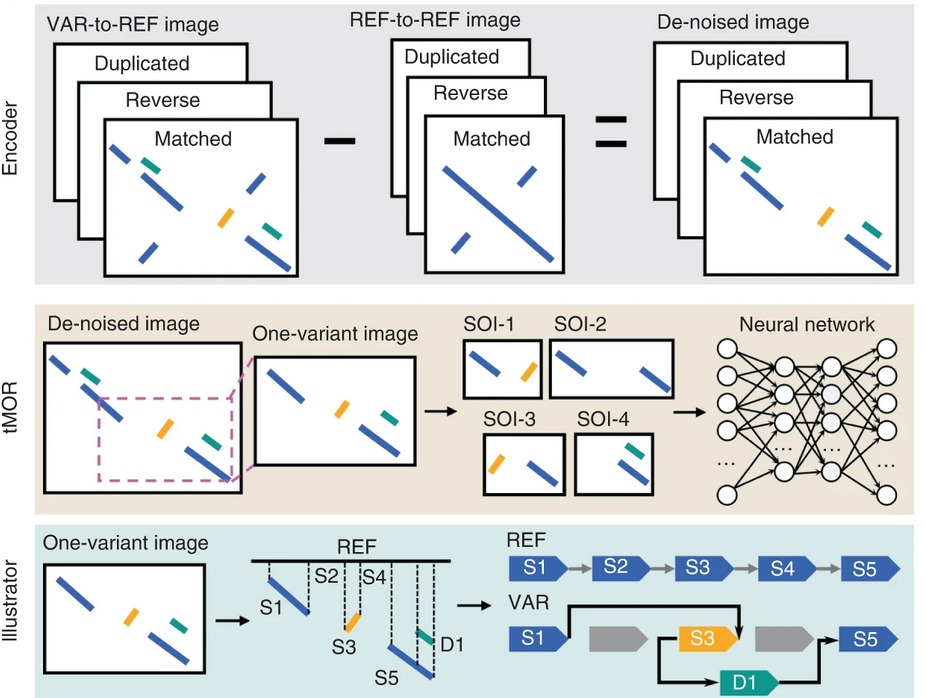
图 20 SVision 流程示意图
图片来源于 Lin et al. SVision: a deep learning approach to resolve complex structural variants. DOI: 10.1038/s41592-022-01609-w
四
总结与展望
变异检测是定位功能基因或疾病基因的基础。如表格 1所示,从测序数据的产生、预处理、基因组的比对、变异检测都可以使用GPU加速来提升检测的速度。高速分析能够节省测序的时间成本,从而降低测序价格和分析成本。这可以让研究人员更愿意进行测序来建立与完善疾病肿瘤相关的数据库,为将来攻克疾病与肿瘤打下基础。
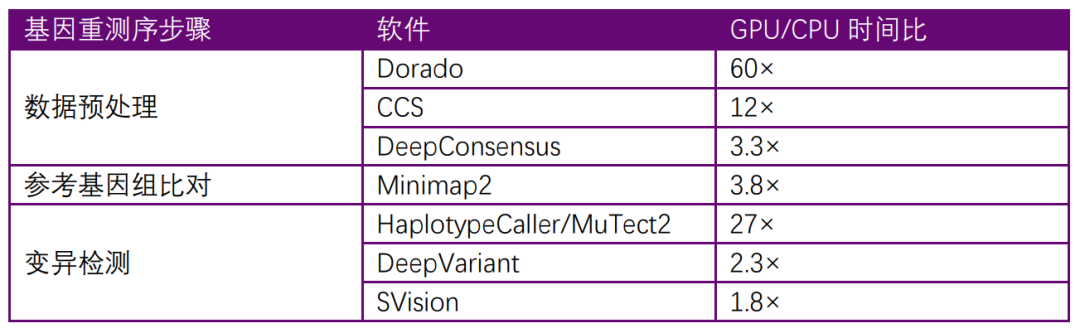
表格 1 文中所提的软件GPU与CPU运行时间比
可以看到,在目前基因测序领域,传统软件算法因为设备局限性,在面对复杂的生物系统时,选择使用简化的概率模型建模来牺牲准确度而获得更快的速度。而随着GPU算力的进步,越来越多研究人员选择深度学习或者大模型来得到更精确的结果。除了文中所提到的数据预处理、基因组比对和变异检测步骤以外,在后续分析中,机器学习也起到了更重要的作用,
比如用线性模型建模关联样本表型与基因型的GWAS可以使用 DeepNull 得到更好的关联结果。
我们预计,随着机器学习算法和GPU设备的发展,快速而准确的基因测序可以帮助研发人员设计靶向药物,帮助普通民众更早发现潜在风险,帮助医生更准确地对病情进行诊断治疗,精准医疗未来可期。
名词列表
责任编辑:彭菁
-
传感器
+关注
关注
2578文章
55875浏览量
795693 -
gpu
+关注
关注
28文章
5335浏览量
136246 -
基因
+关注
关注
0文章
95浏览量
17833
发布评论请先 登录
Xilinx FPGA在基因组测序中的优势
全基因组数据CNV分析简介 精选资料分享
全基因组测序的优势 精选资料分享
什么是基因组序列数据库
全国首个“城区全民基因组测序项目”在深圳启动,基因科技进入临床应用
FPGA能在实时基因组测序计算中大显身手,大大缩短时间
Clay Breshears博士讨论基因组测序和生物信息学
国产芯片助力全球首次实现手机个人全基因组测序分析
国产芯片助力全球首次实现手机个人全基因组测序分析
AI加速推动医疗个体化转型 基因组学将有望成为未来发展主流
人工智能如何改变基因组学?
GPU助力基因组重测序分析



评论